由香港大學(港大)化學系楊軍博士所率領的量子化學研究團隊,最近在英國皇家化學學會的旗艦學術期刊《化學科學》(Chemical Science) 中,發表了一套適用於處理光物理過程的高精度量子化學演算法。此方案具廣泛應用性,適用於研究許多光活性材料的電子及能量轉移通道,對認識材料激發態過程的基礎研究具有重要意義,亦對以高精度和大規模量子數值模擬為基礎的有機光活性材料的設計和優化,提供了新的可能性。
背景介紹
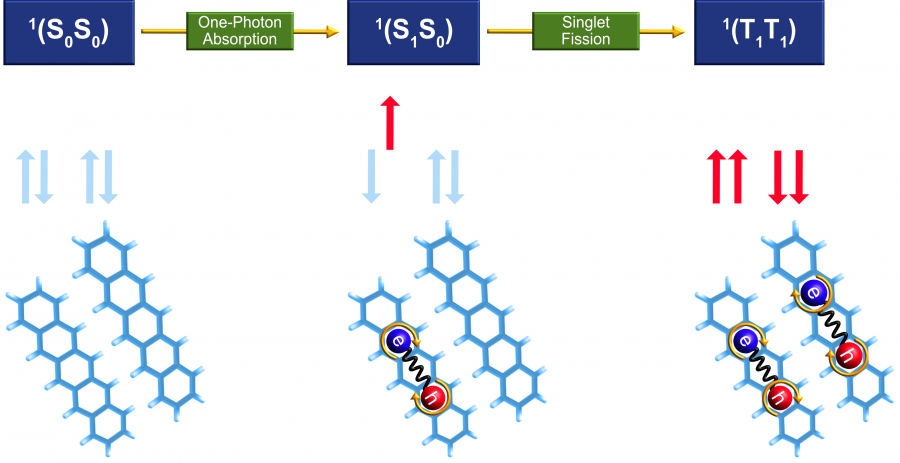
傳統硅基單結太陽能電池的能量轉換效率受到Shockley-Queisser極限的限制(即電池效率受物理性質約束,在正常的陽光條件下,其轉換效率最高為33%),然而在第三代太陽能電池中,可以通過設計新型光伏機制來突破某些傳統光物理過程的限制,進而減少能量損失及提升光電轉換效率。譬如單線態裂分(The singlet fission process,即有機半導體材料受激發產生單線態激子後,通過一個自旋允許的裂分過程,形成兩個三線態激子的多激子產生現象),能將材料吸收的一份光子能量裂分為兩份電子空穴對,理論上光子 - 激子轉換效率會翻倍。通過激子複製實現器件內電流倍增,突破Shockley-Queisser效率上限,從而提高光電轉換效率。
自60年代提出這機制以來,學術界進行了大量的實驗及理論研究,涵蓋基礎理論、材料設計和器件發展。然而由於單線態裂分涉及複雜的光物理過程,一直以來科學家對激子複製和能量損耗通路本性的認識仍存有不少爭議、分歧及概念上的混亂。研究團隊認為,這可歸咎於常規計算方法的理論局限,尚未能準確揭示單線態裂分過程中產生的電子 - 振動態量子多體協同相互作用,因而影響人們對單線態裂分的化學 - 物理關係的理解,限制了裂分材料的設計和開發應用。
由港大開發的複雜量子化學算法,清晰地揭示了並五苯二聚體(Pentacene dimer)單線態的裂分過程,闡明了弱電荷分離態、強電荷分離態、強電子相關三線態對,以及振動量子態等多體相互作用的協同和轉化機制。
研究方法及成就
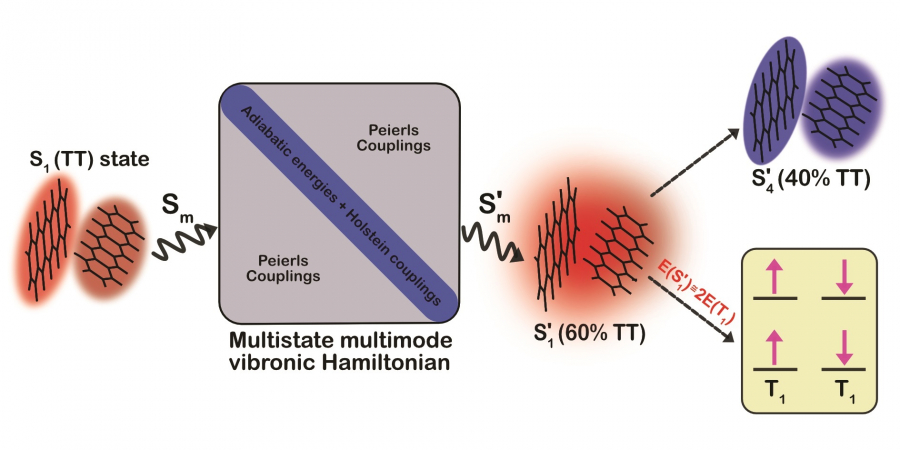
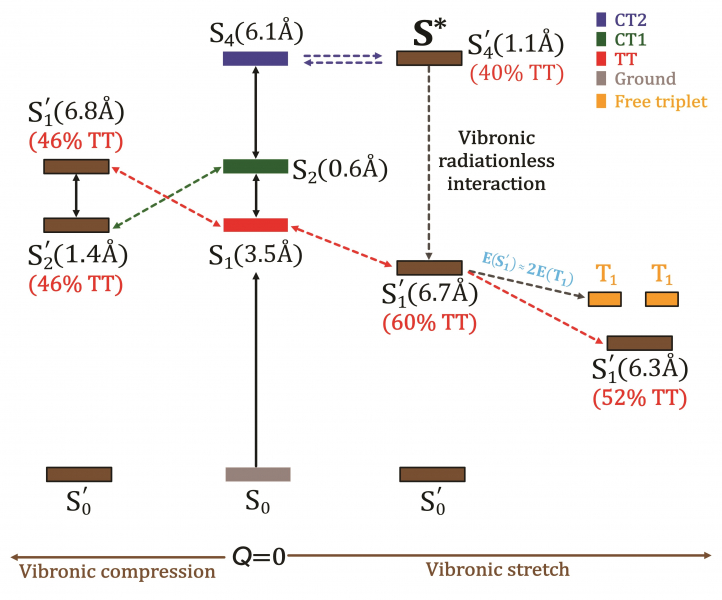
港大團隊提出,單線態裂分過程的精準計算,必須考慮不同性質的分子量子態的多體相互作用,這些量子態可以通過非常精準的數值演算方法「從頭算密度矩陣重整化群自洽場(Ab-initio density matrix renormalisation group self-consistent field method)」算法來獲取。團隊通過對並五苯二聚體的反覆計算和數值分析發現,大量的π介層電子、全部低能單線態以及全部分子振動頻譜的電子-振動耦合作用,對單線態裂分過程至關重要。同時,通過計算和分析量子波函數,可以獲取各激子能態在指定化學結構單元上的局域電子自旋分佈,以及電子自旋態隨二聚體分子振動的演化。這是在單線態裂分領域裏首次發展基於高精度激發態波函數的電子-振動分析,也對構建功能發光材料的化學組分、結構同激發態量子自由度的關聯,提供了新的計算工具。
研究取得的主要成就包括:
1. 發展出一套適用於處理包括單線態裂分在內的光物理過程複雜電子 - 振動耦合量子態的量子化學算法,準確描述量子多體相互作用。
2. 文獻關於並五苯的電線態裂分過程主流結論認為,由電荷分離態的強靜電相互作用來誘導。港大以高精度計算清晰揭示了電荷分離態的本質作用,通過雙重電荷分離態增強電子-分子振動耦合,從而誘導三線態對的活性和去局域化。
楊博士表示:「這項工說明,要產生高效的單線態裂分,過程是非常複雜的,涉及多個量子態,傳統的計算方法掛一漏萬,難以窺其全貌。而港大此次的工作對研究這一類複雜光物理過程發展了系統性的計算工具,令人鼓舞,同時給光電材料提供了新的設計思路,比如可以通過相應地修改化學成分和結構來利用這些電子轉移的路徑。」
本文第一作者、博士生Rajat Walia補充說:「我們正在利用這一算法和研究成果,通過研究含參雜的分子間及分子內單線態裂分機制、增加電荷分離通道以及優化分子間排列等各種方式,以篩選裂分速率較高的材料。」
論文連結:https://doi.org/10.1039/D1SC01703A
關於研究團隊
此項研究由港大化學系助理教授楊軍博士所率領的團隊完成。博士生Rajat Walia是第一作者,楊博士為通訊作者,博士後鄧襗祥博士亦有份參與研究。
關於港大量子化學研究小組的更多資料,請參看:www.junyanglab.com。
本研究得到香港研究資助局、深圳市科技創新計劃和廣東省基礎研究與應用研究重大計劃的資助。團隊也感謝香港創新局AIR@InnoHK創新平台的支持。
相⽚下載及說明:https://www.scifac.hku.hk/press
傳媒如有查詢,請聯絡理學院外務主任杜之樺(電話:3917 4948 ; 電郵:caseyto@hku.hk / 助理傳訊總監陳詩迪(電話:3917 5286 ; 電郵:cindycst@hku.hk)。