
Media
HKU Identifies Novel DNA Changes in Patients with Hirschsprung disease that disrupt the development of the enteric nervous system
26 Aug 2015
Hirschsprung (HSCR) disease (congenital megacolon) is one of the more common birth defects. It is a global problem but is particularly prevalent in Asia, affecting 1 in 3000 babies. These babies suffer from severe constipation and intestinal obstruction because nerve cells which co-ordinate bowel movement are absent. The reasons for the absence of nerve cells in the bowel are unclear, and therefore treatment remains unsatisfactory.
Li Ka Shing Faculty of Medicine, The University of Hong Kong (HKU) has conducted a genetic screen to identify disease-causing DNA changes and established experimental mouse models to investigate the mechanisms underlying and how these DNA changes cause HSCR disease. With the mouse models, the study identified that specific DNA changes in GLI1, GLI2 or GLI3 genes (genes that work coordinately to control the formation of the nerve cells in bowel) disrupt the formation of the nerve cells and their subsequent gut colonisation, resulting in absence of nerve cells at the distal end of the colon as seen in HSCR patients. The novel finding provides important breakthrough for understanding the pathology of this genetic disease. The research has just been published in “Gastroenterology”, the top journal in the field of Gastroenterology.
Leading researcher of the study, Dr Elly Ngan Sau-wai, Associate Professor of the Department of Surgery, Li Ka Shing Faculty of Medicine, HKU remarks, “Better understanding the disease mechanisms will help designing stratified patient care leading to improved outcome in the long run.”
About Hirschsprung’s (HSCR) disease
Babies born with HSCR disease will die unless the portion of the bowel with no nerve cells is surgically removed. In Hong Kong, over 120 affected babies have been operated in the last ten years. Nevertheless, the functional outcome of surgery is variable and a significant number of patients still suffer from life-long complications, ranging from intractable constipation, incontinence, enterocolitis to devastating short bowel syndrome, leading to not only tremendous psychosocial impact on the patients, but also a heavy financial burden to the health care system.
HSCR disease is attributed to a failure in formation of nerve cells in the intestine. During development, neural stem cells in the bowel receive varied signals from their environment and differentiate into a wide-range of nerve cells, which subsequently form a neural network to control the bowel movement. At least two types of cells are present in the bowel, the functional nerve cells (neurons) and the nerve-supporting cells (glia). This ratio of neurons and glia cells has to be precisely regulated, in order to establish a functional network. DNA changes in the genes implicated this process may disturb the formation of functional neural network, leading to HSCR disease.
Study results
Based on the previous findings published in 2009 and 2011, the HKU research team further elucidated how gene batteries that control the formation of nerve cells versus nerve-supporting cells are regulated during development, and how its perturbation causes HSCR disease. The team has performed targeted deep-sequencing and identified several novel mutations in GLI1, 2, 3 in HSCR patients that all result in increased GLI activity, leading to increased SOX10 expression. In mouse, at the early stage of the development, majority of the gut is colonised by neurons and only very few glia can be detected. However, if the mice carry abnormal GLI proteins resulted from loss of SUFU, the mice show premature formation of glia, accompanied by an obvious delay of gut colonisation. The situation is similar to the patient with HSCR disease. Functionally, GLI and SOX10 work coordinately with another GLI regulator, namely SUFU to form a regulatory loop to control formation of nerve cells versus nerve-supporting cells and gut colonisation. Thus, mutations in GLI will interrupt this regulatory network and contribute to HSCR pathogenesis.
Collectively, HKU researchers have identified several novel GLI mutations in HSCR patients. With the functional data, the study directly demonstrated for the first time the coordination of SUFU-GLI-SOX10 gene in formation of nerve cells and nerve-supporting cells in the bowel and that its perturbation leads to HSCR in mouse and human.
Previous study results
In the last decade, the HKU research team has conducted various genetic and functional studies to explore the genetic basis of HSCR. The first whole genome genetic screen was conducted in 2007 on 200 patients with HSCR and 408 healthy individuals to identify the susceptibility genes for HSCR disease. The work was published in “Proceedings of the National Academy of Sciences” in 2009. Subsequently, the team employed another sophisticated computational and statistics methods to identify a discrete number of genes which interact with each other to cause HSCR disease. With the mouse models, the study identified that specific DNA changes in Patched (PTCH1) and Delta-like 3 (DLL3) genes will lead to premature development of nerve-supporting cells, thus increase the risk of having HSCR by 278%. The research was published in “Journal of Clinical Investigation” in 2011, the top journal in the field of developmental biology.
About the HKU research team
This study was performed at Department of Surgery, Li Ka Shing Faculty of Medicine, HKU, and was led by Dr Elly Ngan Sau-wai. The mouse genetic studies were performed by a PhD student, Miss Jessica Liu Aijia and a Postdoctoral fellow, Dr Frank Lai Pui-ling of the Department of Surgery, in collaboration with Professor Hui Chi-chung, Hospital for Sick Children, University of Toronto, Canada. Genetic analysis was performed in collaboration with Dr Maria-Mercedes Garcia-Barcelo and Professor Paul Tam Kwong-hang of Department of Surgery, and Dr Gui Hong-sheng of Department of Psychiatry, HKU. Other researchers include Professor Sham Mai-har of the School of Biomedical Sciences, Li Ka Shing Faculty of Medicine, HKU.
Acknowledgement
This work was supported by a seed funding grant for basic research from the University of Hong Kong, and by research grants (HKU17116914 and T12C-714/14-R) from the Research Grants Council.
Please visit the website at http://www.med.hku.hk/v1/news-and-events/press-releases for press photos.
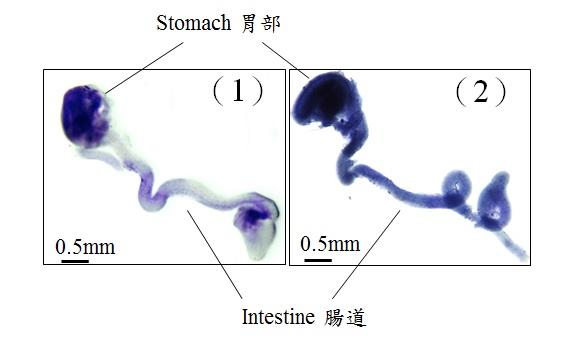
During development, neural stem cells differentiate into a wide-range of nerve cells, which subsequently form a neural network to control the bowel movement. In general, neurogenesis starts first to generate varied types of neurons, which is then followed by gliogenesis, formation of the supporting cells (glia) for the neurons. In the figures, glia is blue in color. 1) At the early stage of the development, only very few glia can be detected in the wild-type mouse gut (light blue); 2) Premature formation of glia was observed in the mutant gut with abnormal GLI protein (dark blue).

In the figures, neurons are green in color. 1) At the early stage of the development, majority of the gut is colonised by neurons; 2) an obvious delay of gut colonisation was observed in mice with abnormal GLI protein, that mimics the situation of patient with Hirschsprung disease. Red line indicates the distance of gut colonisation.